
|
|||
|---|---|---|---|
|
|
Exercício, amônia e sistema nervoso central. Envolvimento dos receptores NMDA |
|
|
|
Universidade Iguaçu (UNIG Itaperuna) Universidade Estácio de Sá (Campos) |
Marco Machado marcomachado1@aol.com (Brasil) |
|
|
|
|
|||
|
|
http://www.efdeportes.com/ Revista Digital - Buenos Aires - Año 10 - N° 89 - Octubre de 2005 |
|
|
1 / 1
Introdução
A amônia é uma molécula envolvida em várias situações metabólicas. Dependendo do pH pode se apresentar como íon (NH4+) ou como gás (NH3). Como o pK da amônia a 37º C é de 9,15, cerca de 98% desta se encontra na forma ionizada nos fluidos fisiológicos (FELIPO e BUTTERWORTH, 2002b; ROSE, 2002). Em qualquer dessas formas a amônia tem livre transito através de membranas celulares e pode ser produto e substrato de diversas reações enzimáticas nas células do SNC (FELIPO e BUTTERWORTH, 2002b; BUTTERWORTH, 2002).
Grandes quantidades de amônia entram no organismo a partir veia porta vinda do sistema gastro-intestinal, contudo, a concentração desta molécula se mantém baixa (50-100 mM) em virtude de um eficiente mecanismo hepático de remoção (FELIPO e BUTTERWORTH, 2002b; BUTTERWORTH, 2002). Falhas neste mecanismo sejam por desordens no sistema metabólico, sejam por lesão hepática, resultará em hiperamonemia e conseqüentemente em uma série de sintomas neuro-psiquiátricos (MONFORT et al, 2000; KELLY e STANLEY, 2001).
A hiperamonemia ter origem congênita ou adquirida. Em sua forma congênita está normalmente relacionada à falhas no ciclo da uréia, mas marcadamente na ornitina transcarbamilase (OTC ou ornitina carbamoil transferase, E.C. 2.1.3.3) responsável pela etapa de conversão de ornitina em citrulina. Já a hiperamonemia adquirida está relacionada à falência hepática provocada por ingestão de toxinas (inclusive etanol), infecções virais ou doenças auto-imunes (FELIPO e BUTTERWORTH, 2002b).
Podemos ainda classificar a hiperamonemia em aguda, geralmente associada à rápida morte de pacientes ou animais, e crônica moderada, associada a alterações na função cerebral (alterações no ciclo sono/vigília, coordenação neuromuscular, cognição, etc.) (MONFORT et al, 2000; ROSE, 2002).
Exercícios até exaustão também podem levar a aumentos na concentração de amônia no córtex, cerebelo e striatum de encéfalos de ratos (GUEZENNEC et al, 1998).
Metabolismo de amônia no SNC (Lançadeira Glutamato/Glutamina)O metabolismo de amônia no SNC está diretamente ligado ao ciclo glutamina/glutamato, essencial para os neurônios glutamatérgicos. Este ciclo envolve a síntese de glutamato no terminal pré-sináptico pela reação de desaminação oxidativa catalisada peta glutamato desidrogenase (GDH, EC. 3.5.1.2), glutamato este liberado na fenda sináptica para transmissão do impulso nervoso (KELLY e STANLEY, 2001; LEBON et al, 2002; FELIPO e BUTTERWORTH, 2002b).
A captação do glutamato se dá principalmente pelos astrócitos através de transportadores, assim que é captado é convertido a glutamina pela glutamina sintetase (GS, E.C. 6.3.1.2) e liberado para o interstício e re-captado pelo neurônio pré-sináptico (LEBON et al, 2002; SUÁREZ et al, 2002; BLÜML et al, 2002) (Figura 1).
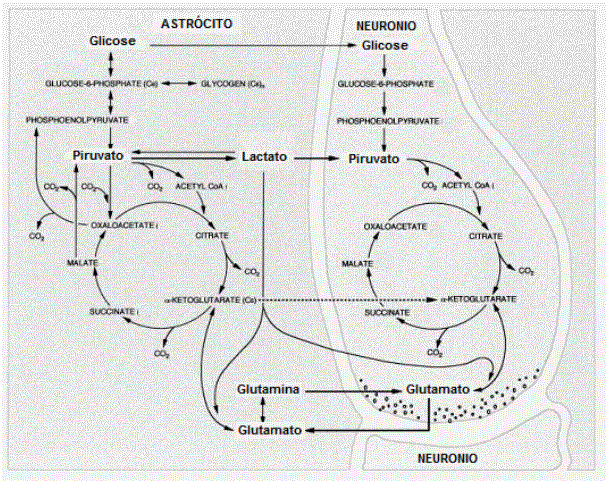
Figura 1 - Integração metabólica no SNC. O glutamato liberado na fenda sináptica é captado principalmente pelos astrócitos e convertido em glutamina pela GS. A glutamina é exportada para o neurônio que a reconverte em glutamato. Praticamente todo o pool de glutamato no SNC é sintetizado a partir do a-cetoglutarato do ciclo do ácido tricarboxilico produzidos no próprio SNC (adaptado de HERTZ et al., 2000).Praticamente todo o pool de glutamato/glutamina em adultos é gerado no próprio SNC, já que estes aminoácidos têm passagem dificultada na barreira hemato-encefálica (HERTZ et al, 2000; BLÜML et al, 2002; ROSE, 2002).
A atividade da GS é fundamental não só para o controle da concentração de amônia como também para manutenção da concentração de glutamato intersticial, prevenindo assim a excitotoxidade1 (SUÁREZ et al, 2002; LYNCH e GUTTMANN, 2002). A síntese de GS é maior nas células gliais próximas dos sítios onde se encontram grandes concentrações de neurônios glutamatérgicos (SUÁREZ et al, 2002).
A relação entre a concentração arterial e cerebral de amônia em condições normais é de aproximadamente 2:1, assim sendo, as células gliais devem estar continuamente captando glutamato e gerando glutamina para que os efeitos tóxicos da amônia não sejam evidenciados (COOPER, 2001; BUTTERWORTH, 2002). A captação da amônia pelo SNC é inversamente proporcional ao fluxo sanguíneo, já que a menor velocidade de fluxo induz mais tempo de contato entre as moléculas de amônia e a barreira hemato-encefálica (FELIPO e BUTTERWORTH, 2002b).
HiperamonemiaA hiperamonemia causa diversos efeitos na função e atividade das células do SNC.
a. Função mitocondrial e metabolismo energético.O metabolismo energético no SNC é alterado de forma significativa, fato que pode ser observado em parte pelo aumento na produção de lactato neste tecido quando submetido a concentrações elevadas de amônia (HERTZ et al, 2000). É marcante também uma maior atividade da fosfofrutoquinase (PFK) e dos transportadores de glicose (GLUT-1). Estes dados sugerem um aumento na atividade glicolítica dos neurônios e quanto maior a concentração de amônia menor é a capacidade dos neurônios em gerar energia. São propostos dois mecanismos para explicar a diminuição na produção de ATP: (a) inibição do ciclo do ácido tricarboxílico (TCA) e (b) mecanismos envolvendo os receptores NMDA (FELIPO e BUTTERWORTH, 2002b).
O aumento da produção de lactato, não permitindo que o piruvato produzido seja conduzido ao TCA, e a inibição da atividade da a-cetoglutarato desidrogenase (aKGDH) reforça a primeira hipótese (COOPER, 2001; FELIPO e BUTTERWORTH, 2002b). Já a segunda hipótese é evidenciada pelo aumento na atividade da bomba Na+K+ATPase provocado pelo desequilíbrio iônico causado pela hiperatividade dos receptores NMDA que funcionam como canais de Ca+2, este aumento na atividade da bomba geraria uma depleção do ATP (COOPER, 2001; FELIPO e BUTTERWORTH, 2002b).
b. Funcionamento dos astrócitos.A exposição dos astrócitos a hiperamonemia provoca modificações morfológicas neste tipo de célula. É comum observar inchamento (swelling) (CHAN et al, 2001) que acarretará numa herniação causando aumento da pressão intracraniana (KELLY & STANLEY, 2001). Além disso, várias proteínas responsáveis pelas funções dos astrócitos são diretamente e indiretamente afetadas pela hiperamonemia.
A glial fibrillarly acidic protein (GFPA) é a principal constituinte dos filamentos intermediários do citoesqueleto dos astrócitos, e tanto o respectivo mRNA quanto à própria proteína são produzidas em menor quantidade nas células expostas a concentrações elevadas de amônia, facilitando assim a desestruturação da célula (FELIPO e BUTTERWORTH, 2002b).
Vários transportadores de aminoácidos têm sua atividade inibida em presença de concentrações elevadas de amônia. Os astrócitos são os principais responsáveis pela remoção do glutamato da fenda sináptica e isto se dá por transportadores de alta afinidade chamados de EAAT-1 (ou GLAST) e EAAT-2 (ou GLT-1) (CHAN et al, 2001). Um terceiro transportador (EAAT-3) encontra-se no neurônio pré-sináptico, porém sua contribuição na re-captação do glutamato é reduzida (ROSE, 2002).
A expressão do mRNA de EAAT-1 e EAAT-2 é menor, tanto in vivo quanto in vitro, quando há exposição à amônia em concentrações aumentadas (BUTTERWORTH, 2001; FELIPO e BUTTERWORTH, 2002B; SUÁREZ et al, 2002). CHAN e colaboradores (2001) demonstraram que a quantidade de EAAT-1 (GLAST) bem como do respectivo mRNA é significativamente menor em astrócitos de ratos expostos a concentrações de amônia maiores do que as normais por 7 dias. A diminuição na quantidade destes transportadores leva a uma manutenção do glutamato durante maior tempo na fenda sináptica aumentando a possibilidade de exitotocicidade (FELIPO e BUTTERWORTH, 2002b).
c. Sistema glutamato.Vários estudos demonstraram que em condições de hiperamonemia ocorre uma diminuição na quantidade de sítios para [3H]Cainato e perda de receptores AMPA, sem alteração ou com aumento dos receptores NMDA (ROSE, 2002). A atividade dos neurônios glutamatérgicos também é diminuída, visto que altas concentrações de amônia tendem a inibir a síntese de glutamato no neurônio pré-sináptico bem como a capacidade de ativação dos receptores AMPA nos neurônios pós-sinápticos (ROSE, 2002).
d. Sistema GABA.O ácido g-aminobutirico (GABA) é um dos principais neurotransmissores inibitórios de SNC. Durante exposição concentrações de amônia entre 0,2-0,5 mM causam aumento da afinidade dos receptores neuronais de GABA(A) numa função semelhante a dos barbituratos (JONES, 2002).
Os transportadores astrocíticos de GABA também sofrem influencia da amônia, em exposições agudas a captação chega a diminuir em 30% e em quatro dias esta captação pode chegar a diminuir em 50-60% (JONES, 2002). Via de transdução de sinal dos receptores NMDA
Ativação dos receptores N-metil-D-aspartato (NMDA) resultam em aumento do Ca+2 intracelular no neurônio pós-sináptico. Ca+2 liga-se a calmodulina e ativa a oxido nítrico sintase (NOS) que por sua vez ativa a guanilato ciclase (GC) aumentando as concentrações de cGMP (Figura 2) (MONFORT et al, 2000; BUTTERWORTH, 2001).
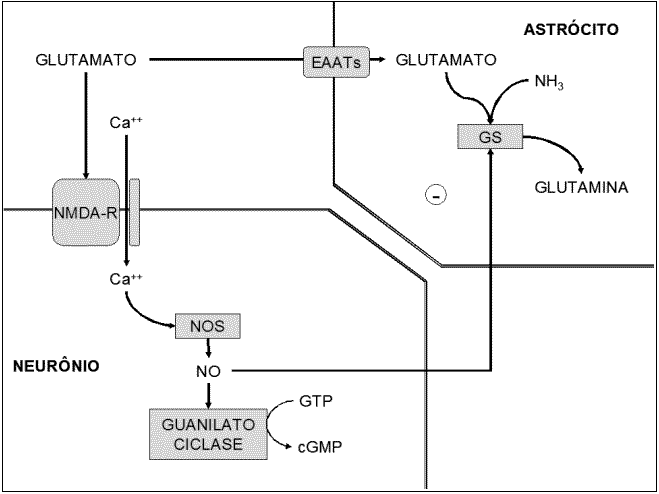
Figura 2 - Via da transdução de sinais do receptor NMDA. Ca+2 liga-se a calmodulina e ativa a oxido nítrico sintase (NOS) que por sua vez ativa a guanilato ciclase (GC) aumentando as concentrações de cGMP. (NMDA-R) receptor NMDA, (EAATs) transportadores de glutamato, (GS) glutamina sintetase.Butterworth (2001) sugere que em estados de hiperamonemia há aumentos de atividade dos receptores NMDA anteriores a aumentos da concentração de glutamato, o que indica que há ligação entre a amônia e o próprio receptor, ativando a via de sinalização.
O aumento excessivo nas concentrações de Ca+2 desdobra-se em várias frentes que podem induzir a morte celular via necrose ou apoptose.
Amônia e exercícioO exercício, como qualquer estresse, altera a demanda energética e por isso o metabolismo. As alterações metabólicas induzem a utilização de reservas e a produção de inúmeros metabólitos que podem ou não ser nocivos ao próprio organismo. Entre estes metabólitos a amônia vem sendo cada vez mais estudada.
Durante o exercício o acompanhamento do comportamento do pool de aminoácidos no sangue periférico pode nos fornecer informações sobre como o metabolismo esta sendo alterado (MacLEAN et al., 1991). Aminoácidos na célula muscular podem ceder o grupamento amina a um cetoácido ou glutamato gerando alanina ou glutamina (respectivamente), que são exportadas via corrente sanguínea e novamente desaminadas no fígado, onde sua cadeia lateral pode servir como agente neoglicogênico e seu grupamento amina para formação de uréia (menos tóxica ao organismo do que a amônia para posterior excreção) (GIBALA et al, 1997; GIBALA et al, 1998; BLOMSTRAND e SALTIN, 1999).
Em alguns casos o processo anaplerótico pode gerar diretamente amônia e esta é exportada via corrente sanguínea (GIBALA et al, 1997; GIBALA et al, 1998). Dependendo do tipo e intensidade da atividade física esta amônia plasmática pode atingir concentrações suficientes para alterar tanto o metabolismo de amônia/neuro-transmissores quanto à atividade excitatória dos neurônios (GUEZENNEC et al, 1998).
Durante atividades físicas, outra fonte de amônia vem da via de degradação de nucleotídeos com aparecimento concomitante de IMP (HELLSTEN et al, 1999; McCONELL et al, 1999; GOREHAM et al, 1999), hipoxantina (HARGREAVES et al, 1998; BALDWIN et al, 1999) e urato (BELLINGER et al, 2000). Quando a demanda energética é alta e a concentração de ADP aumenta, ocorre a ativação da mioquinase (MK), enzima responsável pela transferência de fosfatos entre dois ADP, sendo os produtos ATP e AMP. A relação entre as concentrações de ATP, ADP e AMP é importante na regulação do metabolismo, sendo assim a produção de AMP pela MK deve ser rapidamente equilibrada, isso ocorre a partir da ativação da AMP deaminase (AD) que produz IMP e amônia (figura 3).
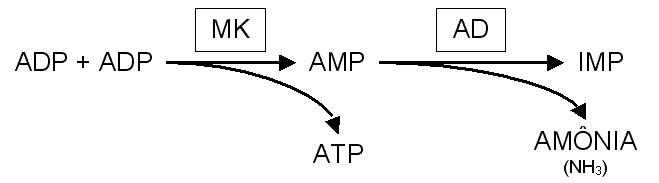
Figura 3 - Gênese da amônia a partir da degradação de adenosina. A ação da mioquinase (MK) transfere um fosfato do ADP para outro com produção de ATP e AMP. O aumento da concentração de AMP ativa a AMP deaminase (AD) que tem como produto o IMP e a amônia (NH3). Estas reações fazem parte do ciclo da adenina nucleotídeo.MacLean et al. (1991) demonstraram que a diminuição da ingestão de carboidratos aumenta a amonemia em relação à ingestão normal, contudo neste estudo a ingestão total de energia foi menor em aproximadamente 55% para o grupo com baixo carboidrato.
Guezennec e colaboradores (1998) demonstraram que a suplementação de creatina diminui a produção de amônia e conseqüentemente aumentou a performance em 60 min de ciclismo. Concomitantemente, houve menor produção de hipoxantina e urato, dado que permitiu aos autores postularem que a suplementação de cretina teve um efeito poupador das purinas.
Fadiga centralDurante muito tempo o modelo aceito para a fadiga central foi o proposto por Newshome. O diminuição da biodisponibilidade dos aminoácidos de cadeia ramificada (BCAA) leva a uma alteração na relação destes com o triptofano o que induziria ao aumento da síntese de serotonina no SNC. A serotonina age como inibidora da atividade neuronal levando a sensação de torpor característica da fadiga (para revisão ver BLOMSTRAND, 2001).
Hoje alguns pesquisadores postulam que a amônia possa ser co-responsável pela fadiga central (BANISTER e CAMERON, 1990; LOPES e CAMERON, 2003). Como citado anteriormente, a hiperamonemia clínica causa efeitos como alterações no ciclo sono/vigília, coordenação neuromuscular, cognição, etc. Estes efeitos são observados temporariamente em indivíduos que apresentam hiperamonemia sub-clinica, ou seja, estados em que a amonemia está elevada mas que não ultrapassam aqueles valores considerados normais. Esta alteração na amonemia é similar à encontrada em exercícios extenuantes e, principalmente naqueles em que a duração é elevada. Isto permite o postulado de que a amônia possa ser mais um dos fatores relacionados com a fadiga central.
ConclusãoMais estudos precisam ser feitos para esclarecer os aspectos que envolvem o exercício e a amonemia, principalmente seus efeitos centrais.
O envolvimento da hiperamonemia sub-clínica afetando o funcionamento do SNC já vem sendo relatado por alguns estudos (HARTMANN et al., 2000; GROENEWEG et al., 2000), porém estes estudos relacionam estados como alcolismo ou hepatite como causadores da hiperamonemia. Estes dados remetem a uma quadro onde a hiperamonemia induzida por exercício possa ser um dos fatores responsáveis para a fadiga central.
A partir daí o envolvimento da atividade física e seus desdobramentos (quantidade, intensidade e modalidade) além de aspectos derivados do status nutricional e cronobiológicos (ciclo circadiano, ciclo menstrual, idade, etc.) podem ser estudados com perspectivas futuras de aplicação prática, sejam no âmbito desportivo como no de lazer ou saúde.
AgradecimentosAo professor L. C. Cameron pelo acesso a informações de seu laboratório e pela iniciação no assunto abordado.
Nota
Injuria neural causada pela não remoção do neurotransmissor, mantendo o neurônio em atividade constante.
Referencias
Baldwin, J.; Snow, R. J.; Carey, M.F. Febbraio, M. A. Muscle IMP accumulation during fatiguing submaximal exercise in endurance trained and untrained men. American Journal of Physiology. v. 277, n. 46, p. R295-R300, 1999.
Banister, E. W.; Cameron, B. J. C. Exercise-induced hyperammonemia: Peripheral and central effects. International Journal of Sports Medicine. v. 11(sup 2), p. 129-142, 1990.
Blomstrand, E. Amino acids and central fatigue. Amino Acids v. 20, p. 25-34, 2001.
Blomstrand, E; Saltin, B. Effect of muscle glycogen on glucose, lactate and amino acid metabolism during exercise and recovery in human subjects. J of Physiology v. 14, p. 293-302, 1999.
Blüml, S.; Moreno-Torres, A.; Shic, F.; Nguy, C.-H.; Ross, B. D. Tricarboxilic Acid Cicle of Glia in the in vivo Human Brain. NMR Biomedicine. v. 15, p. 1-5, 2002.
Butterworth, R. F. Glutamate transporter and receptor function in disorders of ammonia metabolism. Mental Retardation and Developmental Disabilities Research Reviews. v. 7, p. 276-279, 2001.
Butterworth, R. F. Pathophysiology of Hepatic Encephalopathy: A New Look at Ammonia. Metabolic Brain Disease. v. 17, n. 4, p. 221-227, 2002.
Chan, H.; Hazell, A. S. ; Desjardins, P.; Butterworth, R.F. Effects of ammonia on glutamate transporter (GLAST) protein and mRNA in cultured rat cortical astrocytes. Neurochemistry International. v. 37, p. 243-248, 2000.
Cooper, A. J. L. Role of Glutamine in Cerebral Nitrogen Metabolism and Ammonia Neurotoxicity. Mental Retardation and Developmental Disabilities Research Reviews. v. 7, p. 280-286, 2001.
Felipo, V.; Butterworth, R. F. Mitochondrial dysfunction in acute hyperammonemia. Neurochemistry International. v. 40, p. 487-491, 2002a.
Felipo, V.; Butterworth, R. F. Neurobiology of ammonia. Progress in Neurobiology. v. 67, p. 259-279, 2002b.
Gibala, M. J.; MacLean, D. A.; Graham, T. E.; Saltin, B. Anaplerotic processes in human skeletal muscle during brief dynamic exercise. Journal of Physiology. v. 502, p. 3, p. 703-713, 1997.
Gibala, M. J.; MacLean, D. A.; Graham, T. E.; Saltin, B. Tricarboxylic acid cycle intermediate pool size and estimated cycle flux in human muscle during exercise. American Journal of Physiology. v. 275, n. 38, p. E235- E242, 1998.
Goreham, C.; Green, H. J.; Ball-Burnett, M.; Ranney, D. High-resistance training and muscle metabolism during prolonged exercise. American Journal of Physiology. v. 276, n. 39, p. E489-E496, 1999.
Groeneweg, M.; Moerland, W.; Quero, J. C.; Hop, W. C. J.; Krabbe, P. E.; Schalm, S. W. Screening of subclinical hepatic encephalopathy. Journal of Hepatology, 32 : 748-753, 2000.
Guezennec, C.Y.; Abdelmalki, A.; Serrurier, B.; Merino, D.; Bigard, X.; Berthelot, M. Pierard, C.; Peres, M. Effects of prolongued exercise on brain ammonia and amino acids. Int. J. Sports Med., 19 : 323-327, 1998.
Hargreaves, M.; McKenna, M. J.; Jenkins, D. G.; Warmington, S. A.; Li, J. L.; Snow, R. J.; Febbraio, M. A. Muscle metabolites and performance during high-intensity, intermittent exercise. J. Appl. Physiol. 84 (5): 1687-1691, 1998.
Hartmann, I. J. C.; Groeneweg, M.; Quero, J. C.; Beijeman, S. J.; de Man, R. A.; Hop, W. C. J.; Schalm, S. W. The Prognostic Significance of Subclinical Hepatic Encephalopathy. Am. J. Of Gastroenterology, 95(8) : 2029-2034, 2000.
Hellsten, Y.; Richter, E. A.; Kiens, B.; Bangsbo, J. AMP deamination and purine exchange in human skeletal muscle during and after intense exercise. J. of Physiology. 520(3) : 909-920, 1999.
Hertz, L.; Yu, A. C. H.; Kala, G.; Schousboe, A. Neuronal-astrocytic and cytosolic-mitochondrial metabolite traffcking during brain activation, hyperammonemia and energy deprivation. Neurochemistry International, 37 : 83-102, 2000.
Jones, E. A. Ammonia, the GABA Neurotransmitter System, and Hepatic Encephalopathy. Metabolic Brain Disease, 17(4) : 275-281, 2002.
Kanamori, K.; Ross, B. D.; Kondrat, R. W. Glial uptake of neurotransmitter glutamate from the extracellular Fluid studied in vivo by microdialysis and 13C NMR. Journal of Neurochemistry ,83 : 682 -695, 2002.
Kelly, A; Stanley, C. A. Disorders of Glutamate Metabolism. Mental Retardation and Developmental Disabilities Research Reviews, 7 : 287-295, 2001.
Lebon, V.; Petersen, K. F.; Cline, G. W.; Shen, J.; Mason, G. F.; Dufour, S.; Behar, K. L.; Shulman, G. I.; Rothman, D. L. Astroglial Contribution to Brain Energy Metabolism in Humans Revealed by 13C Nuclear Magnetic Resonance Spectroscopy: Elucidation of the Dominant Pathway for Neurotransmitter Glutamate Repletion and Measurement of Astrocytic Oxidative Metabolism. The Journal of Neuroscience, 22(5) : 1523-1531, 2002.
Lopes, L.; Cameron, L. C. Creatina, genese de amônia e o sistema nervoso central. Fitness & Performance Journal, 02(01): 06, 2003
Lynch, D. R.; Guttmann, R. P. Excitotoxicity: Perspectives Based on N-Methyl-D-Aspartate Receptor Subtypes. The Journal of Pharmacology and Experimental Therapeutics. V. 300, n 3, p. 717-723, 2002.
MacLean, D. A.; Spriet, L. L.; Grahan, T. E. Plasma Amino Acid and Ammonia Responses to Altered Diunnetary Intakes Prior to Prolonged Exercise in Humans. Can. J. Physiol. Parmacol. 70 : 420-427, 1991.
McConell, G.; Snow, R. J.; Proietto, J.; Hargreaves, M. Muscle metabolism during prolonged exercise in humans: influence of carbohydrate availability. J. Appl. Physiol. 87(3): 1083-1086, 1999.
Monfort, P.; Kosenko, E.; Erceg, S.; Canales, J. J.; Felipo, V. Molecular mechanism of acute ammonia toxicity: role of NMDA receptors. Neurochemistry International 41 : 95-102, 2002.
Monfort, P.; Montoliu, C.; Hermenegildo, C.; Muñoz, M-D.; Felipo, V. Differential effects of acute and chronic hyperammonemia on signal transduction pathways asociated to NMDA receptors. Neurochemistry International, 37 : 249-253, 2000.
Sáez, R.; Llansola, M.; Felipo, V. Chronic Exposure to Ammonia Alters Pathways Modulating Phosphorylation of Microtubule-Associated Protein 2 in Cerebellar Neurons in Culture. J. Neurochem. 73 : 2555-2562, 1999.
Suárez, I.; Bodega, G.; Fernández, B. Glutamine synthetase in brain: effect of ammonia. Neurochemistry International, 41 : 123-142, 2002.
| |
|
|---|---|
|
revista
digital · Año 10 · N° 89 | Buenos Aires, Octubre 2005 |
|